
-
The first (phase 1) is a dose-finding study: it focuses on tolerability, the evaluation of the maximum tolerated dose, and the identification of the recommended dose to be administered in the subsequent phase (phase 2) of sulfasalazine when combined with standard “3+7” induction chemotherapy, consisting of infusion of idarubicin and cytarabine as described above;
-
The second (phase 2) is intended to confirm the good tolerability of sulfasalazine at the dose recommended for phase 2 when combined with “3+7” chemotherapy, and to preliminarily evaluate the efficacy of adding sulfasalazine to standard “3+7” induction chemotherapy.
If you participate in phase 1 of this study, the dose of sulfasalazine that you will receive will depend on your order of enrollment in the study and on the tolerability of this medicinal product in combination with “3+7” chemotherapy in patients with AML previously treated within the framework of this research.
If you participate in phase 2, the dose of sulfasalazine that you will receive in combination with “3+7” chemotherapy will have been identified at the end of the first phase of this study.
To address the research question, it is planned to include a maximum total of 64 individuals with newly diagnosed AML treated in approximately ten different hematology departments, all located in France.
Please find below some information about your disease that may help clarify the objective of this study:
Acute myeloid leukemia is characterized by the accumulation in the bone marrow and sometimes in the blood of leukemic cancer cells. The other blood cells normally produced by the bone marrow are no longer generated, and the leukemia is responsible for a decrease in red blood cells (anemia), a decrease in platelets (thrombocytopenia, with a risk of bleeding), and a decrease in normal white blood cells, resulting in immune deficiency and leading to infections, often bacterial, which may be severe. Acute myeloid leukemia is a serious disease that, in the absence of treatment, has an unfavorable course within a few months.
Standard induction chemotherapy (“3+7”), independently of the effect of sulfasalazine, leads to transient aplasia corresponding to a cessation of blood cell production by the bone marrow, during which hospitalization in a single room is required. During this period, red blood cell and platelet transfusions are necessary, and the use of antibiotics is frequent. These constraints and treatments are related to the standard induction course and are not imposed by the research.
As you have understood, this induction course often makes it possible to achieve complete remission of your disease.
After your induction course, even if the remission achieved is, as explained above, “complete and deep,” you will need to receive additional cycles of chemotherapy (referred to as consolidation) to prevent AML from returning (relapsing). These consolidation courses may also induce transient aplasia, most often shorter than that following the induction course. It is also possible that you may be offered a bone marrow transplant, most often after a first consolidation cycle. This involves the transfusion of cells derived from the blood or bone marrow of a healthy donor, cells capable of producing all blood components (referred to in medical terminology as an “allogeneic hematopoietic stem cell transplantation”). The donor may be a member of your family, or an unrelated volunteer donor of bone marrow–derived cells. In all cases, these transplanted cells must be genetically sufficiently similar to your own.
In the absence of transplantation, your physician may also propose so-called maintenance treatment with Onureg® tablets, administered 14 days per month. At the beginning of each consolidation chemotherapy cycle or prior to bone marrow transplantation, and then every 3 months during the year following the end of the induction course, a medical visit will take place within the framework of the research in order to collect information on your health status (clinical examination, routine laboratory tests). No additional procedures will be imposed by the research (in particular bone marrow sampling) during this period.
Sulfasalazine will be added only to induction chemotherapy (“3+7”). Furthermore, even if, at the end of this study, this medicinal product were shown to be effective and well tolerated, it would not replace additional treatment with consolidation chemotherapy, possible bone marrow transplantation, and/or maintenance treatment with Onureg®.
One year after the end of your induction chemotherapy, a medical visit will be organized at the time of your study completion. After your study completion, information regarding the status of your disease (remission or relapse) and your outcome will nevertheless be periodically requested by the study organizers from your physician during the 5 years following the end of the research, without you being personally disturbed.
You may find information concerning AML and its treatments on the websites of the French cooperative groups ALFA (www.alfa.leukemia.org) and FILO (FILO-leucemie.org).


{kind=link}
DREAM study
As part of the standard care for your disease or as part of a protocol, your doctor has offered you a treatment based on azacitidine, combined with either venetoclax or ivosidenib depending on the characteristics of your disease, with the aim of achieving remission. The clinical efficacy of these treatments has been established. They are internationally recognized standard treatments for your disease. However, it has been established that even when the first course of treatment is maximally effective, some residual leukemia cells remain, requiring further cycles of treatment after complete remission has been achieved.
The research proposed as part of the DREAM study aims to determine whether it is possible, based on laboratory analysis of blood or bone marrow samples, to predict the effectiveness of treatment, but also to improve biological knowledge about the minority of residual leukemia cells in order to identify ways to improve these treatments.
To do this, the DREAM study relies on new technologies available at the Leukemia Institute, which enable leukemia cells to be cultured in vitro to determine their response to treatment, or to study how diseased cells react to treatment using a blood or bone marrow sample (a technology known as “single-cell sequencing”).
To answer the question posed in the research, this bicentric study with minimal risks and constraints planned to include 120 people newly admitted to the St Louis Hospital and Avicenne Hospital for AML.
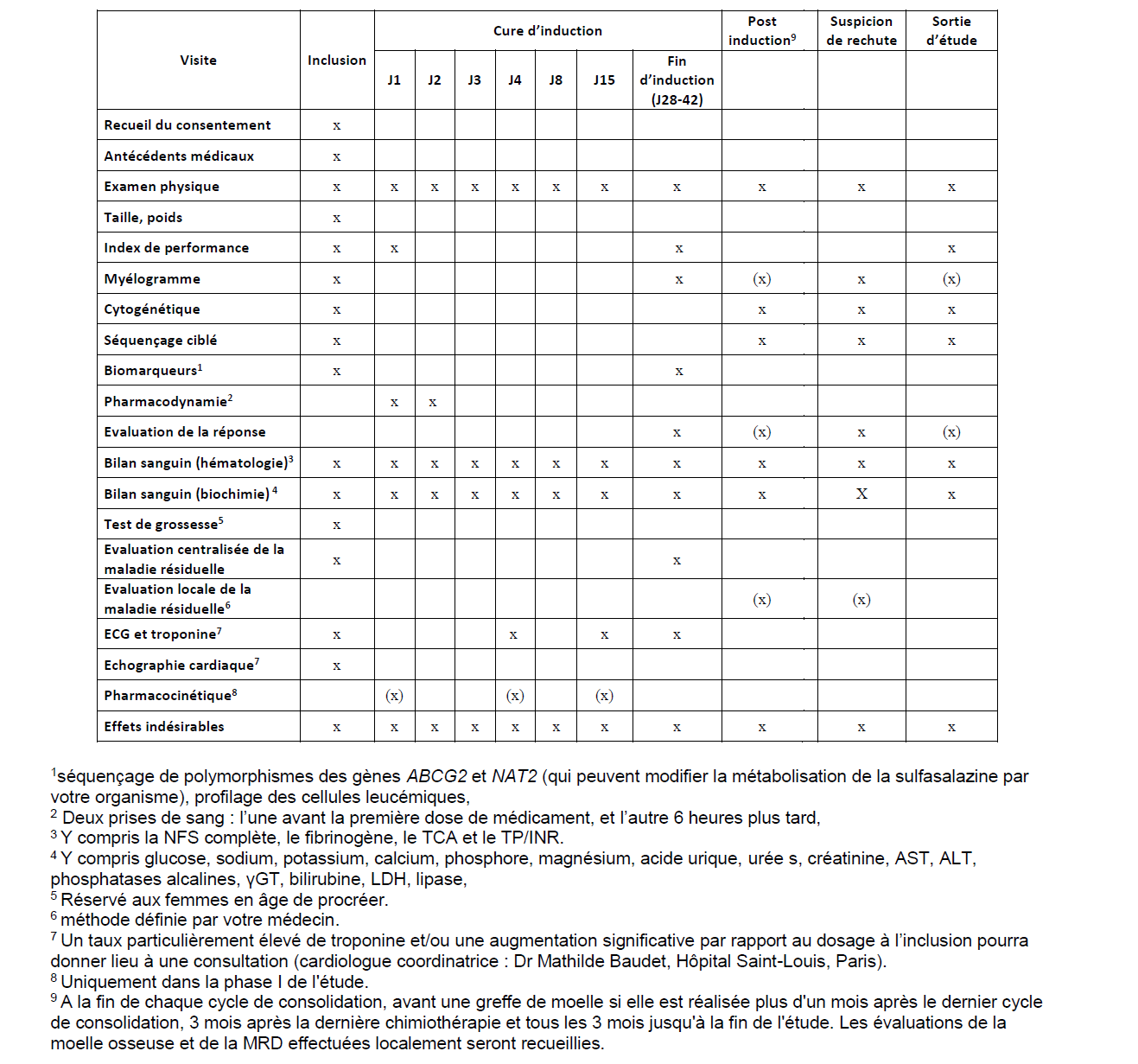
Before starting the first course of treatment, then during the administration period of this first course (4 to 10 days), and finally when evaluating the response to the first and sixth cycles (generally between 21 and 56 days after the start of the cycle), your doctor will prescribe various biological tests (cellular, molecular, protein, and microbiological) of your blood and bone marrow at regular intervals in order to better characterize the disease, monitor its progression during treatment, and monitor the effects of the disease and its treatment on your health.
In the proposed research, we will conduct biological studies on additional blood or bone marrow samples at certain key moments during treatment:
This study aims to validate an in vitro drug response test as a “biomarker” of treatment response. It also aims to refine our understanding of the molecular consequences of these drugs’ action on leukemia cells, and in particular to determine the heterogeneity of response from one cell to another. It is therefore conducted purely for research purposes and will not result in any changes to the treatments your doctor will administer to you. The results of these biological studies will be obtained after your medical treatment has been completed and will have no impact on it.
These additional tests may require up to 6 additional blood tubes (30 mL) to be drawn, most often during a blood draw already scheduled for your monitoring. It is possible that adherence to the study blood draw schedule may result in an additional blood draw. Similarly, an additional 2 mL of bone marrow will be collected during a bone marrow puncture performed at the start of treatment, during the first cycle, and then during the reassessment at the end of the first cycle. While it is standard practice to perform a bone marrow puncture after the first cycle of treatment, an additional intermediate puncture will be performed around the 7th day of treatment as part of this study. This will be crucial in enabling the research physicians to understand the mechanism of action of the treatments, with a view to ultimately improving their efficacy.
Finally, if you give your specific consent, your doctor will perform a biopsy under local anesthesia instead of a simple bone marrow aspiration before starting treatment. This procedure, which is part of the routine tests for certain blood and bone marrow diseases, is generally performed if the aspiration fails.
With your specific consent, this examination will be performed immediately, and if necessary during the reassessment. In addition to the diagnostic information usually obtained with a fine needle aspiration, it will provide information on their spatial organization that only a needle biopsy can provide. New technologies (spatial transcriptomics) will enable medical researchers to better understand how diseased cells are distributed in the bone marrow and with which normal cells in the body (immune system cells, support cells known as “stromal” cells) they interact. This will ultimately lead to the development of new treatment options.
The estimated duration of the study is 38 months, and your participation will be 56 days. After signing your consent form, the study will proceed as follows:
– A 2 mL bone marrow sample will be taken during the initial myelogram assessment of the disease.
– Optionally, if you have specifically consented to it, this myelogram (puncture-aspiration) will be replaced by a puncture-biopsy (“bone marrow biopsy”) under local anesthesia.
– A 30 mL blood sample will be taken before any anti-leukemia treatment, preferably on the same day as the initial myelogram.
– On the evening of Day 1 of the first cycle, between 6 and 12 hours after the first dose of venetoclax (or ivosidenib) was administered (“lysis assessment”),
– On Day 2 of the first cycle, before the second dose of venetoclax (or ivosidenib)
– On the day of remission assessment (between Day 21 and Day 56 after the start of the first cycle).
– Around the 7th day of the cycle (between the 4th and 10th day)
– During the evaluation myelogram of the first and 6th cycles (between the 21st and 56th day after the start of the cycle).
– Optionally, if you have specifically consented to it, this myelogram (aspiration) will be replaced by a biopsy (“bone marrow biopsy”) under local anesthesia.
By participating in this research, you will contribute to a better understanding of how leukemia treatments work.
As part of this academic research, no financial compensation is provided in return for your participation.
The limitations of this study are related to the need to take samples at a fixed time or on a fixed day, and to make it mandatory to perform an intermediate reassessment myelogram. These limitations are minimal because blood samples can usually be taken during the blood tests required for the routine management of your disease, without the need for additional venipuncture, and because the addition of an intermediate evaluation myelogram has no impact on medical management. Blood and bone marrow samples may cause a temporary discomfort at the time of the puncture, as well as a bruise at the puncture site.
If you agree to participate, you must comply with the following points:
After being collected, blood and bone marrow samples will be separated into plasma, cells, and tissue sections (for biopsies). Some of these samples will be cryopreserved at the INSERM U944 unit at Saint-Louis Hospital (Institut de Recherche Saint-Louis) for future use. This biological collection will be reported to the Ministry of Research and the Regional Health Agency (Article L. 1243-3 of the Public Health Code). Another part will be used directly for molecular and/or cellular analyses (RNA and/or DNA sequencing on single cells, flow cytometry, plasma protein assays). Most of these analyses will be conducted at the Hôpital Saint-Louis (Institut de Recherche Saint-Louis).
However, it is possible that certain specific analyses of biological samples or their derivatives (RNA, DNA, proteins, cells, etc.) may be conducted by external academic partners in France, Europe, or elsewhere in the world, or may be outsourced to biotechnology companies in France, Europe, or elsewhere in the world. Where applicable, such shipments will be subject to obtaining an export authorization for biological samples derived from human beings issued by the Ministry of Research, in accordance with Articles R.1235-7 et seq. of the Public Health Code.
At the end of the study, unused samples will be stored for future research on the pathology studied. The biological collection obtained as part of this study will be unique due to the availability of samples obtained during treatment, and not just at diagnosis.
The stored samples and anonymized data will be accessible to THEMA investigators. Access (transfer or assignment) by academic research partners may be requested on the basis of annual calls for projects at the end of this research, and scientific projects must be approved by the DREAM study Steering Committee.
Biological samples will be labeled “THEMA – DREAM biobank” and stored for a maximum period of 25 years (20 years after the end of this research) in the INSERM U944 unit of the Saint-Louis Research Institute. Raw data from correlative biological analyses will be stored securely on the server in the MEARY building at the Saint-Louis Hospital. Secondary data (biomarkers) from this research will be compiled in the eTHEMA study’s clinical data collection and managed by the Clinical Research Unit at Saint-Louis Hospital. This data will be stored for 25 years after the start of the DREAM study.
The DREAM study involves analyzing the somatic genetic characteristics of leukemia cells, excluding any constitutional genetic characteristics, i.e., those that could potentially be passed on to offspring. If a biological analysis of these samples were to suggest the study of transmissible genetic characteristics, you would be asked to provide a new, specific consent form.
You may at any time ask the physician supervising you in the context of the research to destroy these biological samples or to object to any further use of them.
If you do not participate in this study, your treatment will remain unchanged, as will the monitoring of blood tests during treatment and the bone marrow assessment at the beginning and end of the first treatment cycle.
At the end of your participation in the DREAM study, your medical care will continue unchanged, as will your participation in the eTHEMA registry, unless you also decide to end your participation in this registry. Your doctor may decide at any time to end your participation and will explain the reasons for doing so.
As part of the research in which you are being asked to participate, your personal data will be processed by AP-HP, the research sponsor and data controller, in order to analyze the results.
This processing is necessary for the research to be carried out, which is in line with the public interest mission entrusted to AP-HP as a public university hospital.
To this end, your medical data will be sent to the Sponsor or to people or partners acting on its behalf, in France or abroad. This data will be identified by a registration number and labeled “THEMA – DREAM database.” This data may also be sent to French health authorities, under conditions that ensure its confidentiality.
In addition to the sponsor and the investigators at the THEMA center, the only recipients of your coded personal data will be the following two subcontractors:
• The provider of the data collection software, hosted in France (the latter may not distribute or analyze the data).
• The study’s data management and biostatistics department, which is part of the THEMA center.
Your medical data that may be used to document a file for the competent authorities may be transmitted to a manufacturer so that a greater number of patients can benefit from the results of the research. This transmission will be carried out under conditions that ensure confidentiality.
Your data may be used for further research or analyses complementary to this research in collaboration with private or public partners, in France or abroad, under conditions that ensure their confidentiality and the same level of protection as European legislation.
You may object at any time to the further use of your data by the physician who is treating you as part of this research.
Your data will only be stored for as long as is strictly necessary and proportionate to the purpose of the research. It will be stored in the data controller’s information systems for up to ten years after the last publication of the research results.
Your data will then be archived in accordance with current regulations.
The computer file used for this research is implemented in accordance with French regulations (amended Data Protection Act) and European regulations (General Data Protection Regulation – GDPR). You have the right to access, rectify, restrict, and object to the processing of data covered by professional secrecy used in the context of this research. These rights can be exercised by contacting the physician in charge of the research, who is the only person who knows your identity (identified on the first page of this document).
If you decide to stop participating in the research, the data collected prior to this decision will be used in accordance with the regulations and exclusively for the purposes of this research. This is because deleting this data could compromise the validity of the research results. In this case, your data will not be used at any time in the future or for any other research.
If you encounter difficulties in exercising your rights, you can contact the AP-HP Data Protection Officer at the following address: protection.donnees.dsi@aphp.fr, who will be able to explain the remedies available to you through the CNIL. You may also exercise your right to lodge a complaint directly with the CNIL (for more information on this subject, please visit www.cnil.fr).
The AP-HP has taken all necessary measures to conduct this research in accordance with the provisions of the Public Health Code applicable to research involving human subjects.
The AP-HP has taken out insurance (0100518814033 230094) covering its civil liability and that of all parties involved with the company HDI–GERLING through BIOMEDICINSURE, whose address is Parc d’Innovation Bretagne Sud C.P.142 56038 Vannes Cedex.
The AP-HP obtained a favorable opinion from the Committee for the Protection of Persons for this research (Comité de protection des personnes Ile de France III) on September 12, 2023.
Your participation in this research is entirely voluntary. Your decision will not affect the quality of care and treatment you are entitled to expect.
Throughout the research, you may ask your doctor for information about your health and explanations about how the research is being conducted.
You may withdraw from the study at any time without justification, without any impact on the continuation of your treatment or the quality of care you receive, and without any impact on your relationship with your doctor. After withdrawing, you may continue to be treated by the same medical team. In this case, the data collected up to the time of withdrawal will be used for the analysis of the study results.
You may at any time ask the doctor supervising your research to destroy these biological samples or object to their further use.
Your medical file will remain confidential and may only be consulted under the responsibility of the doctor treating you, as well as by the health authorities and persons duly authorized by the AP-HP for research purposes and subject to professional secrecy.
At the end of the study and after analysis of the data relating to this study, you may be informed of the overall results by asking the doctor who is treating you as part of this study.
You may also access all of your medical data directly or through a doctor of your choice in accordance with the provisions of Article L 1111-7 of the Public Health Code.
After reading all this information, discussing all aspects with your doctor, and taking time to reflect, if you agree to participate in the research, you will be required to sign and date the informed consent form at the end of this document.