- L’institut
- Un nouvel Institut Hospitalo-Universitaire
- Notre actualité
- Nous soutenir
- Vous êtes
- Patients et proches
- Être soigné et accompagné
- Devenir patient expert
- Découvrir l’Institut
- Chercheurs
- Recherche
- Essais cliniques
- Découvrir l’Institut
- Professionnels de la santé
- Adresser un patient
- Notre recherche clinique
- Découvrir l’Institut
- Industriels
- Découvrir l’Institut
- Recherche translationnelle
- Donateurs
- Nous soutenir
- Découvrir l’Institut
- Soin
- Prise en charge des patients
- Leucémies aiguës myéloïdes
- Leucémies aiguës lymphoïdes
- Néoplasies myéloprolifératives
- Syndromes myélodysplasiques
- Leucémies myéloïdes chroniques
- Nos services cliniques
- Nos laboratoires de biologie médicale
- Informations pour les patients
- Soins de support
- Diagnostic
- Traitements anti-cancéreux
- Recherche
- Nos équipes de recherche
- Nos plateformes technologiques
- Notre recherche clinique sur les leucémies
Portail de transparence
La recherche clinique à l'Institut de la Leucémie

Votre médecin a détecté des anomalies dans les résultats de votre prise de sang correspondant à une quantité trop importante de globules blancs (hyperleucocytose) et/ou une baisse de certains globules (polynucléaires neutrophiles, taux d’hémoglobine reflétant les globules rouges, ou plaquettes). Ces anomalies ont motivé votre consultation ou votre hospitalisation car elles ont fait suspecter une maladie du sang (hémopathie). Au terme des premières investigations, votre médecin a porté le diagnostic de Leucémie et vous avez consenti à participer au registre prospectif de l’Institut de la Leucémie pour mieux comprendre l’origine de ces hémopathies et suivre leur évolution au cours des différents traitements qui peuvent être proposés.
L’institut de la Leucémie en chiffres
1ercentre de lutte contre la leucémie en France
10services cliniques
16équipes de recherche
4laboratoires de biologie médicale
Les services cliniques
Service d’Hématologie et immunologie pédiatrique, Jean-Hugues Dalle
Hôpital Robert-Debré
48 boulevard Sérurier 75019 Paris
Service Adolescents Jeunes Adultes, Nicolas Boissel
Hôpital Saint-Louis
1 avenue Claude Vellefaux 75010 Paris
Service Hématologie Clinique, Olivier Hermine
Hôpital Cochin-Port Royal
27 rue du Faubourg Saint Jacques – 75014 Paris
Service Hématologie Clinique et Thérapie Cellulaire, Thorsten Braun
Hôpital Avicenne Université Sorbonne Paris Nord
125 Route de Stalingrad 93000 Bobigny
Service hématologie-greffe, Régis Peffault de Latour
Hôpital Saint-Louis
1 avenue Claude Vellefaux, 75010 Paris
Service Hématologie Adultes, Emmanuel Raffoux
Hôpital Saint-Louis
1 avenue Claude Vellefaux 75010 Paris
Unité ambulatoire d’hémato-oncogénétique, Marie Sébert
Hôpital Saint-Louis
1 avenue Claude Vellefaux 75010 Paris
Etude DYNHAEMICS
Votre médecin a détecté des anomalies dans les résultats de votre prise de sang correspondant à une quantité trop importante de globules blancs (hyperleucocytose) et/ou une baisse de certains globules (polynucléaires neutrophiles, taux d’hémoglobine reflétant les globules rouges, ou plaquettes). Ces anomalies ont motivé votre consultation ou votre hospitalisation car elles ont fait suspecter une maladie du sang (hémopathie). Au terme des premières investigations, votre médecin a porté le diagnostic de Leucémie Aiguë Myéloïde (LAM) et vous avez consenti à participer au registre prospectif du Centre National de Médecine de Précision (Programme THEMA) dont s’est doté l’Hôpital Saint-Louis pour mieux comprendre l’origine de ces hémopathies et suivre leur évolution au cours des différents traitements qui peuvent être proposés.
Dans le cadre des soins habituels de votre maladie ou dans le cadre d’un protocole, votre médecin vous a proposé un traitement combinant deux médicaments de chimiothérapie (un médicament de la famille des anthracyclines : daunorubicine ou idarubicine, combiné à l’aracytine) dans le but d’obtenir une rémission de la maladie. En fonction des caractéristiques de votre maladie ou de votre participation à un protocole, ces médicaments de chimiothérapie peuvent vous être administrés sous la forme d’injections intraveineuses séparées, ou sous une forme combinée dite « liposomale » (CPX-351, Vyxeos®). L’efficacité clinique de ces traitements, est parfaitement établie et constitue un traitement de référence internationalement reconnu de votre maladie. Pour autant, il est établi que, même en cas d’efficacité maximale de cette première cure de chimiothérapie (obtention d’une rémission complète), quelques cellules leucémiques résiduelles persistent, nécessitant la poursuite des traitements après l’obtention d’une rémission complète.
La recherche proposée dans le cadre de l’étude DYNHAEMICS a pour seul objectif d’améliorer les connaissances biologiques sur cette minorité de cellules leucémiques résiduelles, afin d’identifier en leur sein des talons d’Achille sur lesquels de nouveaux médicaments pourraient à l’avenir être efficaces. Pour cela, l’étude DYNHAEMICS s’appuie sur des nouvelles technologies disponibles à l’Institut de Recherche Saint-Louis, et qui permettent d’étudier cellule par cellule les conséquences moléculaires de la chimiothérapie (technologie dite de « séquençage sur cellules uniques »).
Pour répondre à la question posée dans la recherche, cette étude bicentrique à risques et contraintes minimes a prévu d’inclure 200 personnes nouvellement prises en charge dans les services de l’hôpital St Louis et de l’Hôpital Avicenne pour une LAM.Avant d’initier la première cure de chimiothérapie, puis pendant la période d’administration de cette chimiothérapie (5 à 7 jours), et enfin à l’évaluation de la rémission complète (en règle entre 28 et 56 jours après l’initiation du traitement), votre médecin va être amené à prescrire à intervalles réguliers différentes analyses biologiques (cellulaires, moléculaires, protéiques et microbiologiques) du sang et de la moelle osseuse afin de mieux caractériser la maladie, de suivre son évolution pendant le traitement, et de surveiller les conséquences de la maladie et de son traitement sur votre santé.
Dans la recherche proposée, nous allons conduire des études biologiques sur un volume de sang ou de moelle osseuse supplémentaire à certains moments clés du traitement par chimiothérapie (juste avant son initiation, dans les premières heures suivant son administration, les 2ème , 3ème et 8ème jours du protocole, puis à la sortie d’aplasie et à l’évaluation de la réponse de cette première cure de chimiothérapie).
Cette étude vise à affiner à l’échelle moléculaire la connaissance des conséquences moléculaires de l’action des médicaments de chimiothérapie sur les cellules leucémiques et notamment de déterminer leur hétérogénéité d’une cellule à l’autre. Elle est donc conduite purement à visée de recherche et n’entraîne aucune modification des traitements que votre médecin vous administrera. Les résultats de ces études biologiques seront obtenus de façon différée par rapport à votre prise en charge médicale et n’auront aucune incidence sur celle-ci.
Ces analyses supplémentaires peuvent nécessiter qu’un maximum de 4 tubes de sang supplémentaires (20 ml) soient prélevés, le plus souvent au cours d’une prise de sang déjà prévue pour votre surveillance. Il est possible que le respect du calendrier des prélèvements de l’étude conduise à la réalisation d’un prélèvement sanguin supplémentaire. Celui-ci pourra, sauf cas exceptionnel, être réalisé sur la voie veineuse centrale (cathéter), sans nécessiter de ponction veineuse supplémentaire. De même, 2 ml de moelle osseuse supplémentaires seront prélevés au cours d’une ponction de moelle osseuse effectuée à l’initiation du traitement, en cours de chimiothérapie, puis lors de la réévaluation. S’il est habituel de pratiquer la ponction de moelle osseuse intermédiaire au 15ème jour après l’initiation du traitement, cette ponction intermédiaire sera réalisée le 8ème jour de traitement dans le cadre de cette étude, à la place de l’analyse du 15ème jour. Votre participation à cette étude ne conduira donc pas à la réalisation d’un nombre de ponctions médullaires plus important que pour les patients pris en charge en dehors de cette étude.La durée prévisionnelle de la recherche est de 4 années et 90 jours et votre participation sera de 90 jours. Après la signature de votre consentement, le déroulement de la recherche sera le suivant :
- Dès le bilan d’évaluation initiale de la maladie seront pratiqués :
– un prélèvement de moelle osseuse de 2 ml, au cours du myélogramme d’évaluation initiale de la maladie
– Le médecin qui vous prend en charge pourra vous proposer la réalisation dans le même temps, sous anesthésie locale, d’une biopsie ostéo-médullaire
– Un prélèvement sanguin de 20 ml sera réalisé avant toute introduction de traitement d’attente par hydroxyurée, au mieux le même jour que le myélogramme initial. - Un nouveau prélèvement sanguin de 20 ml sera ensuite réalisé
– Le matin même du début de la chimiothérapie,
– Le soir du Jour 1 de chimiothérapie, entre 6 et 12 heures après l’heure d’administration de la 1ère dose (« bilan de lyse »),
– Les matins des Jours 2, 3 de la cure de chimiothérapie
– Le jour de la sortie d’aplasie (entre le 15ème et le 20ème jours après le début de la cure)
– Le jour de l’évaluation de la rémission (entre le 28ème et le 56ème jour après le début de la cure). - Un nouveau prélèvement médullaire de 2 mL sera ensuite réalisé
– Le matin du Jour 8 de la chimiothérapie
– Au cours du myélogramme d’évaluation de la rémission (entre le 28ème et le 56ème jour après le début de la cure).
En participant à cette recherche, vous contribuerez à une meilleure compréhension du mode d’action des chimiothérapies dans les leucémies.
Dans le cadre de cette recherche académique, aucune compensation financière n’est prévue en contre partie de votre participation.Les contraintes de cette étude sont liées à la nécessité de réaliser les prélèvements de l’étude à heure ou jour fixe, et de rendre impératif la réalisation du myélogramme de réévaluation intermédiaire. Ces contraintes sont minimes car les prélèvements veineux peuvent être réalisés le plus souvent sur la voie veineuse centrale, sans nécessité de ponction veineuse, et que l’anticipation de la date du myélogramme d’évaluation intermédiaire n’a aucune incidence sur la prise en charge médicale. Les prélèvements sanguins et médullaires peuvent entrainer une sensation désagréable transitoire au moment de la ponction, ainsi qu’un hématome au point de ponction.
Si vous acceptez de participer, vous devrez respecter les points suivants :
– Venir aux rendez-vous. En cas d’impossibilité, nous vous remercions de contacter votre médecin le plus rapidement possible.
– Suivre les recommandations de votre médecin relatif à votre participation à l’étude.
– Informer le médecin de la recherche, de l’utilisation de tout traitement ainsi que de tout événement survenant pendant la recherche (hospitalisation, grossesse, prélèvement sanguin réalisé au cours du dernier mois,…).
– Etre affilié(e) à un régime de sécurité sociale ou être bénéficiaire d’un tel régime.Après avoir été prélevés, les échantillons de sang et de moelle vont être fractionnés en plasma et cellules. Une partie de ces échantillons seront cryoconservés dans l’unité INSERM U944 à l’Hôpital Saint-Louis (Institut de Recherche Saint-Louis) pour une utilisation différée. Cette collection biologique sera déclarée au Ministère de la Recherche et à l’Agence Régionale de Santé (Article L. 1243-3 du Code de la Santé Publique). Une autre sera utilisée directement pour des analyses moléculaires et/ou cellulaires (séquençage d’ARN et/ou d’ADN sur cellules uniques, cytométrie en flux, dosage de protéines plasmatiques). La plupart de ces analyses seront conduites sur le site de l’Hôpital Saint-Louis (Institut de Recherche Saint-Louis). Néanmoins, il est possible que certaines analyses spécifiques à partir des échantillons biologiques ou de leurs dérivés (ARN, ADN, protéines, cellules, etc.) soient conduites par des partenaires académiques extérieurs, en France, en Europe ou ailleurs dans le monde, ou soient soumises à des prestations de service à des entreprises de biotechnologies en France, en Europe ou ailleurs dans le monde. Le cas échéant, ces envois seront conditionnés par l’obtention d’une autorisation d’export d’échantillons biologiques issus de la personne humaine émises par le Ministère de la Recherche, en accord avec les Articles R.1235-7 et suivants du Code de la Santé Publique.
Au terme de l’étude, les échantillons non utilisés seront conservés pour des recherches ultérieures sur la pathologie étudiée. En effet, la collection biologique obtenue dans le cadre de cette étude sera unique du fait de la disponibilité de prélèvements obtenus en cours de traitement, et non pas seulement au diagnostic.
Les échantillons conservés et les données anonymisées, seront accessibles aux investigateurs THEMA. Leur accès (cession ou transfert) par des partenaires de recherche académique pourra être sollicité sur la base d’appels à projets annuels au terme de la présente recherche, et les projets scientifiques devront avoir été approuvés par le Comité de Pilotage de l’étude DYNHAEMICS.
Les échantillons biologiques seront labélisés « biobanque – DYNHAEMICS- Fondation ARC » et stockés pour une durée maximale de 25 ans (20 ans après le terme de la présente recherche) dans l’unité INSERM U944 de l’Institut de Recherche Saint-Louis. Les données brutes issues des analyses biologiques corrélatives seront stockées de façon sécurisée sur le serveur du bâtiment MEARY de l’Hôpital Saint-Louis. Les données secondaires (biomarqueurs) issues de ces recherches seront regroupées au sein du recueil de données de cliniques de l’étude eTHEMA et gérées par l’Unité de Recherche Clinique de l’Hôpital Saint-Louis. Ces données seront conservées pendant 25 ans après le début de l’étude DYNHAEMICS.
La réalisation de l’étude DYNHAEMICS prévoit l’analyse des caractéristiques génétiques somatiques des cellules leucémiques, à l’exclusion de toute caractéristique génétique constitutionnelle, c’est-à-dire potentiellement transmissible à la descendance. Si une analyse biologique sur ces échantillons devait proposer l’étude de caractéristiques génétiques transmissibles, un nouveau consentement dédié vous sera soumis.
Vous avez la possibilité à tout moment de demander au médecin qui vous suit dans le cadre de la recherche, la destruction de ces prélèvements biologiques ou de vous opposer à toute utilisation ultérieure.En cas de non-participation à cette étude, votre traitement demeurera inchangé, de même que la surveillance des analyses sanguines en cours de traitement, l’évaluation de la moelle osseuse au début et à la fin de la première cure de chimiothérapie, et votre médecin pourra être amené à vous proposer la réalisation d’une ponction de moelle osseuse intermédiaire autour du 15ème jour après le début du traitement.
A la fin de votre participation à l’étude DYNHAEMICS, votre prise en charge médicale continuera de façon inchangée, ainsi que votre participation au registre eTHEMA, sauf si vous mettez également un terme à votre participation à ce registre. Votre médecin pourra décider à tout moment de l’arrêt de votre participation ; il vous en expliquera les raisons.
Dans le cadre de la recherche à laquelle il vous est proposé de participer, un traitement de vos données personnelles va être mis en oeuvre par l’AP-HP, promoteur de la recherche, et responsable de traitement, pour permettre d’en analyser les résultats.
Ce traitement est nécessaire à la réalisation de la recherche qui répond à la mission d’intérêt public dont est investie l’AP-HP en tant qu’établissement public de santé hospitalo-universitaire.
A cette fin, les données médicales vous concernant seront transmises au Promoteur ou aux personnes ou partenaires agissant pour son compte, en France ou à l’étranger. Ces données seront identifiées par un numéro d’enregistrement et labélisées « base de données – DYNHAEMICS – Fondation ARC ». Ces données pourront également, dans des conditions assurant leur confidentialité, être transmises aux autorités de santé françaises.
Outre le promoteur et les investigateurs du centre THEMA, les seuls destinataires de vos données personnelles codées seront les deux sous-traitants suivants :
– Le fournisseur du logiciel de recueil de données, hébergé en France (celui-ci ne pourra ni les diffuser ni les analyser).
– Le service de gestion des données et de biostatistiques de l’étude, qui fait partie du centre THEMA.
Les données médicales vous concernant pouvant documenter un dossier auprès des autorités compétentes pourront être transmises à un industriel afin qu’un plus grand nombre de patients puissent bénéficier des résultats de la recherche. Cette transmission sera faite dans les conditions assurant leur confidentialité.
Vos données pourront être utilisées pour des recherches ultérieures ou des analyses complémentaires à la présente recherche en collaboration avec des partenaires privés ou publics, en France ou à l’étranger, dans des conditions assurant leur confidentialité et le même niveau de protection que la législation européenne.
Vous pouvez vous opposer à tout moment à l’utilisation ultérieure de vos données auprès du médecin qui vous suit dans le cadre de cette recherche.
Vos données ne seront conservées que pour une durée strictement nécessaire et proportionnée à la finalité de la recherche. Elles seront conservées dans les systèmes d’information du responsable de traitement jusqu’à dix ans après la dernière publication des résultats de la recherche.
Vos données seront ensuite archivées selon la réglementation en vigueur.
Le fichier informatique utilisé pour cette recherche est mis en oeuvre conformément à la règlementation française (loi « Informatique et Libertés » modifiée) et européenne (Règlement Général sur la Protection des Données -RGPD). Vous disposez d’un droit d’accès, de rectification, de limitation et d’opposition au traitement des données couvertes par le secret professionnel utilisées dans le cadre de cette recherche. Ces droits s’exercent auprès du médecin en charge de la recherche qui seul connaît votre identité (identifié en première page du présent document).
Si vous décidez d’arrêter de participer à la recherche, les données recueillies précédemment à cet arrêt seront utilisées conformément à la réglementation, et exclusivement pour les objectifs de cette recherche. En effet, leur effacement serait susceptible de compromettre la validité des résultats de la recherche. Dans ce cas, vos données ne seront absolument pas utilisées ultérieurement ou pour une autre recherche.
En cas de difficultés dans l’exercice de vos droits, vous pouvez saisir le Délégué à la Protection des données de l’AP-HP à l’adresse suivante : protection.donnees.dsi@aphp.fr, qui pourra notamment vous expliquer les voies de recours dont vous disposez auprès de la CNIL. Vous pouvez également exercer votre droit à réclamation directement auprès de la CNIL (pour plus d’informations à ce sujet, rendez-vous sur le site www.cnil.fr ).L’AP-HP a pris toutes les mesures pour mener cette recherche conformément aux dispositions du Code de la Santé Publique applicables aux recherches impliquant la personne humaine.
L’AP-HP a souscrit une assurance (N°0100518814033 210127°) garantissant sa responsabilité civile et celle de tout intervenant auprès de la compagnie HDI–GERLING par l’intermédiaire de BIOMEDICINSURE dont l’adresse est Parc d’Innovation Bretagne Sud C.P.142 56038 Vannes Cedex.
L’AP-HP a obtenu l’avis favorable du Comité de Protection des Personnes (C.P.P.) Sud-Ouest et Outre-Mer 1 pour cette recherche le 15 /11/2021.participation à cette recherche est entièrement libre et volontaire. Votre décision n’entraînera aucun préjudice sur la qualité des soins et des traitements que vous êtes en droit d’attendre.
Vous pourrez tout au long de la recherche demander des informations concernant votre santé ainsi que des explications sur le déroulement de la recherche au médecin qui vous suit.
Vous pouvez vous retirer à tout moment de la recherche sans justification, sans conséquence sur la suite de votre traitement ni la qualité des soins qui vous seront fournis et sans conséquence sur la relation avec votre médecin. A l’issue de ce retrait, vous pourrez être suivi par la même équipe médicale. Dans ce cas, les données collectées jusqu’au retrait seront utilisées pour l’analyse des résultats de la recherche.
Vous avez la possibilité à tout moment de demander au médecin qui vous suit dans le cadre de la recherche, la destruction de ces prélèvements biologiques ou de vous opposer à toute utilisation ultérieure.
Votre dossier médical restera confidentiel et ne pourra être consulté que sous la responsabilité du médecin s’occupant de votre traitement ainsi que par les autorités de santé et par des personnes dûment mandatées par l’AP-HP pour la recherche et soumises au secret professionnel.
A l’issue de la recherche et après analyse des données relatives à cette recherche, vous pourrez être informé(e) des résultats globaux en le demandant au médecin qui vous suit dans le cadre de cette recherche
Vous pouvez également accéder directement ou par l’intermédiaire d’un médecin de votre choix à l’ensemble de vos données médicales en application des dispositions de l’article L 1111-7 du Code de la Santé Publique.
Après avoir lu toutes ces informations, discuté tous les aspects avec votre médecin et après avoir bénéficié d’un temps de réflexion, si vous acceptez de participer à la recherche vous devrez signer et dater le formulaire de consentement éclairé se trouvant à la fin de ce document.Etude SALMA
Vous êtes potentiellement éligible à cette étude parce que vous avez 60 ans ou plus et venez récemment d’être diagnostiqué d’une leucémie aigüe myéloïde (LAM) traitable par une chimiothérapie intensive standard.
L’étude de vos cellules leucémiques a montré qu’elles n’étaient pas porteuses d’anomalies de gènes qui vous aurait rendu éligible à des médicaments particuliers en plus de la chimiothérapie.
Vous devez recevoir une chimiothérapie intensive standard. Celle-ci débute par une cure dite « d’induction » selon un schéma « 3+7 », c’est-à-dire combinant 3 jours de traitement par des perfusions brèves d’une chimiothérapie appelée idarubicine et 7 jours de traitements par une perfusion continue de cytarabine (aussi appelée aracytine).
La cure d’induction vise à obtenir la rémission complète, c’est-à-dire l’absence de cellules leucémiques visibles lors de l’examen de votre moelle osseuse au microscope et le fonctionnement à nouveau normal de votre moelle osseuse. Même lorsqu’elles ne sont plus détectables au microscope, de rares cellules leucémiques résiduelles peuvent persister dans le sang et dans la moelle osseuse. Il faut alors des examens de laboratoire plus élaborés (cytométrie en flux, biologie moléculaire) pour les énumérer. Ces rares cellules leucémiques persistantes forment ce que l’on appelle la « maladie résiduelle ». Leur élimination nécessite la poursuite des traitements après la cure de chimiothérapie d’induction.
La recherche à laquelle nous vous proposons de participer porte sur l’ajout à la chimiothérapie d’induction standard « 3+7 » d’un médicament appelé sulfasalazine. La recherche vise à évaluer chez des patients de 60 ans ou plus et récemment diagnostiqués d’une LAM si l’ajout de ce médicament (sulfasalazine) est sans danger, et s’il augmente le taux de rémission complète profonde, c’est-à-dire d’une rémission complète associée à une maladie résiduelle dite « négative » (cellules leucémiques indétectables ou inférieures à 0,1% des cellules analysées par cytométrie en flux).
L’obtention d’une rémission complète avec maladie résiduelle négative présage souvent d’une rémission prolongée.La sulfasalazine n’est pas une nouvelle molécule. Ce médicament est utilisé (et dispose d’un enregistrement) depuis plus de 40 ans dans le traitement d’autres pathologies (certains rhumatismes inflammatoires et des maladies inflammatoires chroniques de l’intestin). Dans le cadre de la recherche qui vous est proposée, la sulfasalazine s’administre par voie orale, sous forme de comprimés qui doivent être pris 2 à 4 fois par jour pendant 8 à 15 jours.
De récents travaux de recherche ont montré que la sulfasalazine provoquait la mort des cellules de LAM et, en outre, augmentait l’effet anti-leucémique de chimiothérapies de type 3+7, notamment de la classe des anthracyclines (comme l’idarubicine). Le mécanisme d’action de la sulfasalazine a pu en être caractérisé avec précision. Ces travaux ont été présentés dans des congrès médicaux internationaux et font l’objet d’une publication dans un journal scientifique. Si vous avez des connaissances en biologie cellulaire n’hésitez pas à poser des questions sur ces travaux au médecin qui vous propose cette étude.
La recherche à laquelle nous vous proposons de participer comporte successivement deux phases :
- La première (phase 1) est une recherche de dose : elle porte sur la tolérance, l’évaluation de la dose maximale tolérée, et l’identification de la dose recommandable devant être administrée à la phase suivante (phase 2) du médicament sulfasalazine lorsqu’il est combiné avec la chimiothérapie standard d’induction « 3+7 », associant perfusion d’idarubicine et cytarabine comme indiqué ci-dessus ;
- La seconde (phase 2) permet de confirmer la bonne tolérance de la sulfasalazine à la dose recommandée pour la phase 2 lorsqu’elle est combinée à la chimiothérapie « 3+7 », et d’évaluer de façon préliminaire l’efficacité de l’ajout de la sulfasalazine avec la chimiothérapie standard d’induction « 3+7 ».
Si vous participez à la phase 1 de cette étude, la dose de sulfasalazine qui vous sera administrée dépendra de votre ordre d’entrée dans l’étude et de la tolérance de ce médicament en combinaison avec la chimiothérapie « 3+7 » chez les patients atteints de LAM précédemment traités dans le cadre de cette recherche.
Si vous participez à la phase 2, la dose de sulfasalazine qui vous sera administrée en combinaison avec la chimiothérapie « 3+7 » aura été identifiée à la fin de la première phase de cette étude.
Pour répondre à la question posée dans la recherche, il est prévu d’inclure au total un maximum de 64 personnes présentant une LAM récemment diagnostiquée traités dans une dizaine de services d’hématologie différents, tous localisés en France.
Vous trouverez ci-dessous quelques explications sur votre maladie susceptibles d’éclairer l’objectif de cette étude :
La leucémie aigüe myéloïde est l’accumulation dans la moelle osseuse et parfois dans le sang de cellules cancéreuses leucémiques. Les autres cellules du sang normalement fabriquées par la moelle osseuse ne sont plus produites, et la leucémie est responsable de la baisse des globules rouges (anémie) de la baisse des plaquettes (thrombopénie, avec un risque de saignement) et de la baisse des globules blancs normaux responsables d’un déficit immunitaire à l’origine d’infections souvent bactériennes qui peuvent être sévères. La leucémie aigüe myéloïde est une maladie grave qui en l’absence de traitement est d’évolution défavorable en quelques mois.La chimiothérapie standard d’induction (« 3+7 ») conduit, indépendamment de l’effet de la sulfasalazine, à une aplasie transitoire qui correspond à l’arrêt de la production des globules du sang par la moelle osseuse, pendant laquelle une hospitalisation en chambre seule est requise. Pendant cette période, des transfusions de globules rouges et de plaquettes sont nécessaires et il est fréquent d’avoir recours à des antibiotiques. Ces contraintes et ces soins sont liés à la cure d’induction standard et ne sont pas imposées par la recherche.
Comme vous l’avez compris, cette cure d’induction permet souvent d’obtenir une rémission complète de votre maladie.
Après votre cure d’induction, même si la rémission induite est comme expliqué plus haut, « complète et profonde » vous devrez recevoir des cycles supplémentaires de chimiothérapie (dite alors de consolidation) pour éviter que la LAM ne revienne (ne « rechute »). Ces cures de consolidation peuvent induire elles aussi des aplasies transitoires, le plus souvent plus courtes que celle qui fait suite à la cure d’induction. Il est aussi possible que l’on vous propose de recevoir une greffe de moelle le plus souvent après un premier cycle de consolidation. Il s’agit de la transfusion de cellules issues du sang ou de la moelle osseuse d’un donneur en bonne santé, cellules capables de fabriquer tous les éléments du sang (on parle en langage médical de « greffe allogénique de cellules souches hématopoïétiques »). Le donneur peut appartenir à votre famille, ou ne pas être apparenté mais être un volontaire pour le don des cellules issues de la moelle osseuse. Dans tous les cas ces cellules greffées devront être génétiquement suffisamment semblables aux vôtres. En l’absence de greffe, il est également possible que votre médecin vous propose de recevoir un traitement dit « d’entretien » (ou « de maintenance ») par des comprimés d’Onureg®, 14 jours par mois. A chaque début de cycle de chimiothérapie de consolidation ou avant la greffe de moelle, puis tous les 3 mois pendant l’année suivant la fin de la cure d’induction, une visite médicale aura lieu dans le cadre de la recherche pour recueillir des informations sur votre état de santé (examen clinique, examens biologiques de routine). Aucune procédure supplémentaire ne sera imposée par la recherche (notamment prélèvement de moelle osseuse) pendant cette période.
La sulfasalazine ne sera ajoutée qu’à la chimiothérapie d’induction (« 3+7 »). En outre, même si, au terme de cette étude, ce médicament s’avérait efficace et bien toléré, il ne dispenserait pas des traitements complémentaires par chimiothérapie de consolidation, éventuellement greffe de moelle, et/ou traitement d’entretien par Onureg®.
Un an après la fin de votre chimiothérapie d’induction, une visite médicale sera organisée à l’occasion de votre sortie d’étude. Après votre sortie d’étude, des informations sur le statut de votre maladie (rémission ou rechute) et votre devenir seront cependant ponctuellement demandés par les organisateurs de la recherche à votre médecin pendant les 5 ans suivant la fin de la recherche, sans que vous ne soyez personnellement dérangé.
Vous pouvez retrouver des informations concernant la LAM et ses traitements sur les sites des groupes coopérateurs français ALFA (www.alfa.leukemia.org) et FILO (FILO-leucémie.org).
Dans la recherche proposée, nous allons évaluer la sulfasalazine commercialisée dans ses autres indications sous forme de comprimés de 500 mg (par exemple sous le nom de marque Salazopyrine®).
- Si vous participez à la phase 1 de la recherche, vous recevrez selon votre ordre d’entrée dans l’étude et la tolérance à la sulfasalazine observée chez les patients ayant commencé leur participation à l’étude avant vous : soit 1 comprimé 2 fois par jour de J1 à J8, soit 1 comprimé 3 fois par jour de J1 à J8 ; soit 1 comprimé 3 fois par jour J1 à J15 ; soit 2 comprimés 3 fois par jour de J1 à J15 ; soit 3 comprimés 3 fois par jour de J1 à J15 ; soit 4 comprimés 3 fois par jour de J1 à J15 ; soit 4 comprimés 4 fois par jour de J1 à J15.
- Si vous participez à la phase 2 de la recherche, vous recevrez la dose quotidienne et la durée de traitement sélectionnés à la fin de la phase 1 par les organisateurs de la recherche.
- Dans tous les cas, vous recevrez toujours la même dose pendant toute la durée du traitement. Celui-ci pourra être interrompu ou définitivement arrêté par votre médecin investigateur en cas d’effet indésirable.
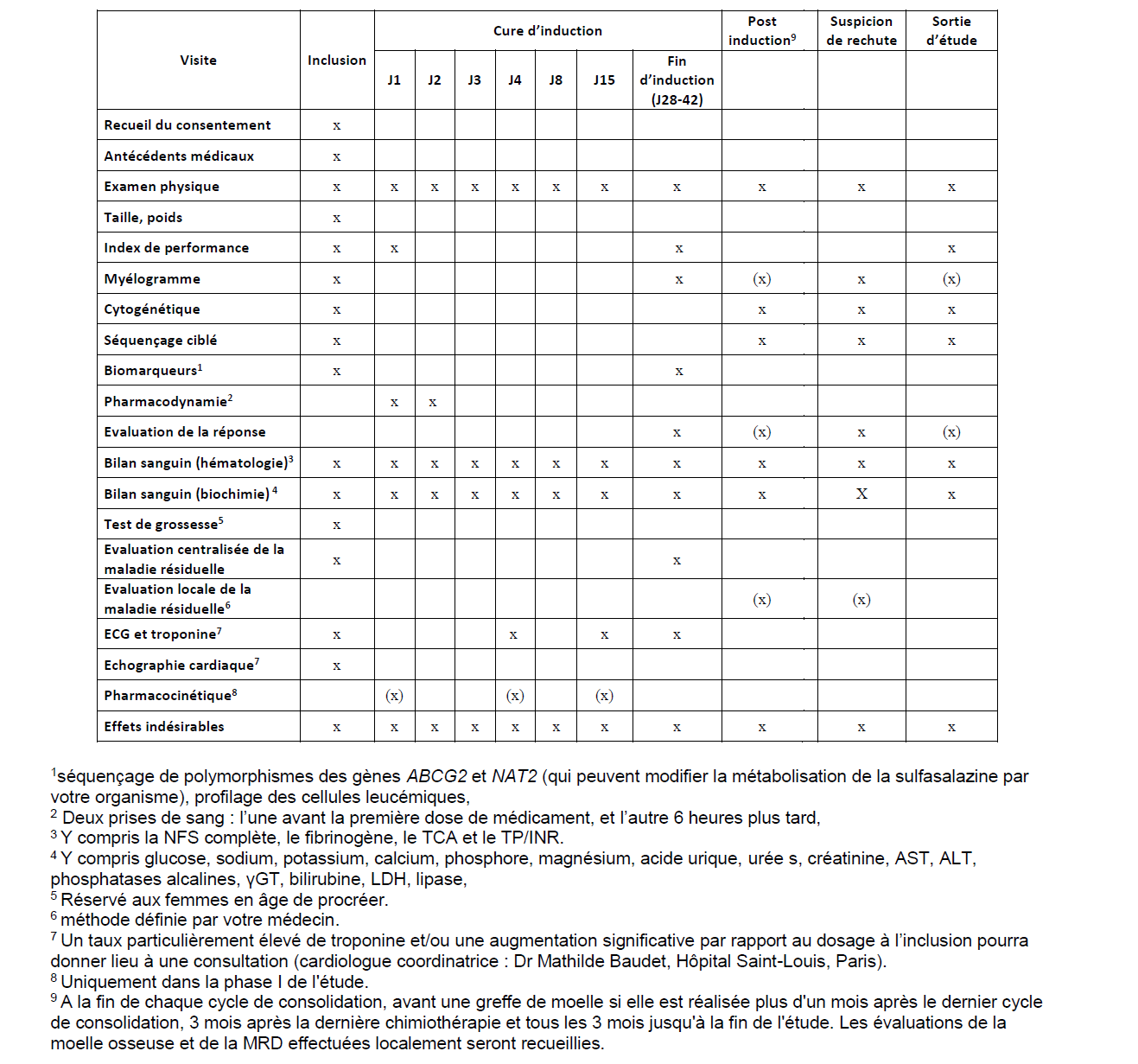
L’évaluation de la sécurité et de l’efficacité du traitement repose sur votre surveillance médicale, des prises de sang, des aspirations de moelle osseuse (myélogrammes) et sur des électrocardiogrammes (« ECG »), comme mentionné dans les sections 3 et 6 de cette note d’information.
La plupart de ces examens seront réalisés alors que vous êtes hospitalisés et dans la cadre de la surveillance habituelle d’une chimiothérapie d’induction « 3+7 ». Après l’induction, et comme indiqué au chapitre 6, la recherche n’imposera que quelques prises de sang et un ECG supplémentaire.
La durée prévisionnelle de la recherche est d’environ 3 ans et votre participation sera de 14 mois.
Après la signature de votre consentement, le déroulement de la recherche sera le suivant :
Phase de sélection
Si vous décidez de participer à cette étude et que vous signez et datez le formulaire de consentement éclairé, votre médecin vous demandera une série d’examens qui seront les suivants :- Un examen clinique,
- Un électrocardiogramme (ECG) et une échographie cardiaque,
- Des prises de sang, la plupart réalisées dans le cadre d’un suivi de routine.
- Une ponction-aspiration de moelle osseuse (myélogramme) pour évaluer les caractéristiques biologiques de votre LAM (analyses des chromosomes et des gènes de la leucémie aigüe myéloïde, typage immunologique de vos cellules leucémiques). Un volume supplémentaire de 2 ml de moelle osseuse est utilisé dans le cadre de la recherche.
Phase d’hospitalisation et de traitement
Vous serez hospitalisé pendant plusieurs semaines lors de votre cure d’induction, et jusqu’à ce que vous sortiez d’aplasie. Au cours de ces hospitalisations seront réalisés une ou plusieurs fois par semaine, les examens suivants :- La liste des événements indésirables que vous avez ressentis,
- La liste des médicaments que vous prenez ou que vous avez pris récemment,
- Un examen clinique,
- Le contrôle de vos signes vitaux (pression artérielle, température),
- Un ECG dans le cadre de la surveillance de routine à J1 et dans le cadre de la recherche à J4, à J15 et à la visite de fin d’induction
- Des prises de sang. La plupart sont réalisées dans la cadre de la surveillance de routine d’une cure de chimiothérapie d’induction « 3+7 » (contrôle de vos fonctions biologiques, biochimiques et de coagulation), mais un volume supplémentaire est prélevé dans la cadre de la recherche (surveillance cardiaque par le dosage de troponine à J1, J4, J15 et à la visite de fin d’induction, 5mL à chaque fois). En outre, des prises de sang supplémentaires seront réalisées à J1 (45 mL en phase I, 10 mL en phase II), J2 (10mL en phase I et II), J4 (45 mL en phase I) et J15 (30 mL, pour la phase I uniquement) pour la recherche (dosages de la sulfasalazine, et évaluation de ses effets biochimiques).
Une prise de sang et une ponction et aspiration du liquide de votre moelle osseuse devront en outre être réalisées entre J28 et J42, après la sortie d’aplasie, c’est-à-dire lorsque votre numération des neutrophiles est ≥ 1 G/L et celle des plaquettes est ≥ 100 G/L, ou à J42 en l’absence de reconstitution hématologique, afin d’évaluer la réponse au traitement et l’évolution de votre LAM. Un volume supplémentaire de moelle osseuse (2 mL) sera alors prélevé pour la recherche.
Visites de suivi après la cure d’induction
En fonction des résultats de l’évaluation de votre moelle osseuse à la fin de la cure d’induction, votre médecin pourra vous proposer d’être ré-hospitalisé pour les cures de consolidation et/ou pour la greffe de cellules souches hématopoïétiques le cas échéant. L’aplasie après chimiothérapie de consolidation et l’aplasie post-greffe sont habituellement plus courtes que l’aplasie post-induction.Une visite de suivi de l’étude intervient à la fin de chaque cycle de consolidation, quand la reconstitution de votre sang et votre maladie sont réévaluées, puis avant la greffe, si celle-ci est réalisée plus d’un mois après la dernière cure de consolidation, enfin en consultation tous les 3 mois jusqu’à votre visite de fin d’étude. La visite de fin d’étude a lieu un an après votre fin de cure d’induction. Une visite peut être planifiée par votre médecin à tout moment s’il suspecte la survenue d’une récidive de la maladie (rechute).
Ce calendrier de suivi n’impose aucune hospitalisation ou visite supplémentaire par rapport à la routine.
Au cours de ces visites, le médecin référent dans l’étude ou un membre de son équipe pourra effectuer les actes suivants afin d’évaluer l’effet du médicament à l’étude et/ou de contrôler votre état de santé :
- Lister les événements indésirables que vous avez ressentis,
- Réaliser un examen clinique,
- Contrôler vos signes vitaux (pression artérielle, température),
- Réaliser des prélèvements sanguins (contrôle de vos fonctions biologiques, biochimiques et de coagulation) et lorsque l’investigateur le juge utile, une ponction aspiration de moelle, étudié localement.
Après votre sortie d’étude, des informations minimales sur le statut de votre maladie (rémission ou rechute) et votre devenir pourront être ponctuellement demandées par les organisateurs de la recherche à votre investigateur, pendant 5 ans, sans que vous ne soyez personnellement dérangé.
Les informations données ci-dessus sont indicatives. Le traitement et sa surveillance peuvent à tout moment être modifiés du fait de l’évolution de votre LAM et de l’apparition de complications.
Le bénéfice attendu du traitement étudié, la sulfasalazine, en addition à la chimiothérapie standard d’induction, est une augmentation de la probabilité d’entrée en rémission profonde de la LAM (rémission complète avec maladie résiduelle négative) et donc de vos chances de survie prolongée.
Même la dose de sulfasalazine la plus faible évaluée dans ce protocole au cours de sa première phase est susceptible d’avoir une activité anti-leucémique.
Par ailleurs en participant à cette étude vous aiderez à accroitre les connaissances sur les traitements de la LAM et pourrez également aider d’autres personnes ayant la même maladie que vous.
Certains médicaments ne sont pas autorisés :
- Les traitements anti-leucémiques autres que ceux décrits dans la rubrique 3 de cette note ;
- Les facteurs de croissance des globules rouges (érythropoïétine recombinante) ou les médicaments stimulant la production des plaquettes (analogues de la thrombopoïétine)
Certains médicaments sont déconseillés sauf si votre investigateur en considérait l’utilisation comme absolument nécessaire à vos soins :
- Les médicaments de la famille des « analogues puriques », notamment l’azathioprine (Imurel®)
- La digoxine
- Les résines échangeuses d’ions (comme le polystyrène sulfonate de sodium, Kayexalate® ou la cholestyramine, Questran®)
Les risques ajoutés par la recherche sont ceux liés à la toxicité potentielle de la sulfasalazine. Ils sont bien connus, compte tenu de la mise sur le marché ancienne de ce médicament, et inconstants :
- La fréquence et l’intensité des évènements indésirables les moins rares augmentent avec la dose utilisée : fatigue, manque d’appétit, nausées voire vomissements, maux de tête. Les nausées et vomissements seront prévenus par l’administration d’anti-nauséeux, utilisés systématiquement pour accompagner la chimiothérapie.
- Rarement et indépendamment de la dose, des réactions dites d’« hypersensibilité » peuvent être observées : une rougeur cutanée, parfois associée à de la fièvre, à l’apparition de ganglions et à diverses anomalies détectables par une prise de sang. Exceptionnellement, des réactions inflammatoires sévères, voir menaçantes pour la vie ont été observées. Elles sont susceptibles d’affecter de multiples tissus et organes. Une telle réaction pourrait survenir alors que vous êtes soumis à une surveillance régulière clinique et biologique par prise de sang ; elle serait rapidement diagnostiquée et conduirait à l’interruption des prises de sulfasalazine.
- De façon hypothétique, la sulfasalazine, par un mécanisme d’action similaire à celui affectant les cellules leucémiques, pourrait augmenter le risque de toxicité cardiaque d’un des médicaments utilisés dans la chimiothérapie standard d’induction : l’idarubicine, justifiant, outre l’échographie cardiaque réalisée avant la mise en route du traitement, une surveillance fréquente de votre coeur par ECG et prise de sang (dosage d’une enzyme, la troponine). Si nécessaire, votre médecin pourra avoir recours aux conseils d’un cardiologue spécialisé dans la prise en charge des problèmes cardiaques des patients cancéreux.
La plupart des contraintes induites par cette étude sont celles des traitements d’une LAM par une chimiothérapie standard d’induction. Les contraintes supplémentaires, limitées à la cure d’induction, spécifiquement liées à l’étude sont les suivantes :
- L’administration quotidienne par voie orale de sulfasalazine pendant les 8 à 15 premiers jours de votre participation à la recherche.
- Si vous participez à l’étude pendant sa phase 1, des prélèvements sanguins de petit volume, mais répétés plusieurs fois par jour pour surveiller l’évolution des concentrations sanguines de sulfasalazine (« pharmacocinétique ») à J1, J4 et J15. Dans les 2 phases de l’essai, du sang sera également prélevé à 2 reprises, à 6 heures d’intervalle, à J1 et J2 et utilisé pour doser diverses substances témoignant du mécanisme d’action de sulfasalazine (« pharmacodynamie »).
- La surveillance de la tolérance cardiaque de sulfasalazine par un dosage sanguin de la troponine à J1, J4, J15 et lors de l’évaluation de fin de cycle d’induction, entre J28 et J42, et par un ECG supplémentaire (non systématiquement réalisé dans la routine de la surveillance des chimiothérapies standards d’induction), à J4 et J8.
Enfin, un volume supplémentaire de 2 mL de moelle sera prélevé lors du myélogramme d’inclusion et lors de la visite de fin d’induction (entre J28 et J42) pour mesurer la cible de la sulfasalazine sur les cellules leucémiques, et différents marqueurs d’état de fonctionnement des cellules leucémiques (« biomarqueurs »). Si vous signez le consentement dédié, une analyse génétique des variations (polymorphismes) de deux gènes qui peuvent influencer la façon dont votre corps assimile la sulfasalazine sera également réalisée.
Cette étude n’ajoute donc aucune hospitalisation supplémentaire ou visites de consultation autres que celles réalisées habituellement dans le cadre d’une chimiothérapie standard d’induction.
Si vous acceptez de participer, vous devrez respecter les points suivants :- Venir aux rendez-vous. En cas d’impossibilité, nous vous remercions de contacter votre médecin le plus rapidement possible.
- Ne pas prendre part à un autre projet de recherche sans l’accord de votre médecin.
- Etre affilié(e) à un régime de sécurité sociale ou être bénéficiaire d’un tel régime.
Les prélèvements de moelle osseuse seront adressés au laboratoire d’hématologie de l’Hôpital Saint-Louis pour l’analyse de la maladie résiduelle par cytométrie en flux, réalisée dans le cadre des soins courants.
Deux types de prélèvements sont utilisés dans cette recherche, dirigés dans plusieurs laboratoires centraux :
- Des prélèvements de sang dirigés vers :
- Le laboratoire de pharmacologie de l’hôpital Saint-Louis à Paris pour les études de pharmacocinétique,
- L’unité de recherche INSERM U944 de l’Institut de Recherche Saint-Louis pour les études de pharmacodynamie.
- Des prélèvements par ponction-aspiration de moelle osseuse confiés à l’unité de recherche INSERM U944 de l’Institut de Recherche Saint-Louis pour les études de pharmacodynamie et de biomarqueurs.
Cela permettra aux organisateurs de la recherche, d’étudier l’effet de la sulfasalazine sur votre maladie et votre organisme. Ces résultats seront exclusivement destinés à des objectifs de recherche. Dans la mesure où ces tests sont de nature exploratoire, ils n’auront aucune conséquence particulière sur vous-même ou sur les affections médicales de votre famille.
Certains échantillons préparés à partir des prélèvements réalisés à votre entrée dans l’étude et lors de l’évaluation de la réponse au traitement, à la fin du cycle d’induction seront conservés congelés dans l’unité INSERM U944 et placés sous la responsabilité du Pr Raphaël Itzykson. Au terme de la recherche, et après déclaration au Ministère de la Recherche et à l’autorité sanitaire régionale compétente (Article L. 1243-3 of the Code de la Santé Publique), ils pourront être conservés pendant une durée maximale de 20 ans puis seront détruits. Ils pourront être utilisés pour des recherches sur la LAM pour répondre à des questions ou en utilisant des techniques qui émergeront avec l’avancée des connaissances et qui ne peuvent être précisées aujourd’hui.
Les résultats des tests ne vous seront pas communiqués de façon systématique. Par contre, à votre demande, une consultation spécifique avec le médecin référent dans l’étude pourra être prévue pour vous faire part des résultats de ces analyses s’ils sont disponibles au moment de votre demande ainsi que les résultats globaux de la recherche lorsqu’ils seront disponibles.
Dans le cas où vous décideriez de ne plus participer à l’étude et retireriez votre consentement, vous pourrez demander, par écrit, que vos prélèvements sanguins ne soient pas analysés dans le cadre de cette étude. Les prélèvements sanguins et médullaires seront alors détruits selon les procédures locales en vigueur.
Les résultats de ces études pourront être publiés dans un ouvrage ou une revue médicale ou être utilisés à des fins pédagogiques. Toutefois vos données resteront confidentielles, ni votre nom, ni aucune information qui pourrait permettre de vous identifier directement ne seront utilisés dans une publication ou un support éducatif.
Vos données et votre matériel biologique pourront faire l’objet de recherches ultérieures ou d’analyses complémentaires, en collaboration avec les autorités compétentes, des partenaires privés ou publics, en France ou dans d’autres pays, y compris hors de l’Union Européenne dans des conditions assurant leur confidentialité et le même niveau de protection que la législation européenne. Vos données et votre matériel biologique peuvent par exemple être utilisés pour le développement futur de nouveaux traitements, même après la fin de cette étude. Ces données et ce matériel peuvent être valorisés par les organisateurs de la recherche. Vous pouvez vous opposer à tout moment à l’utilisation ultérieure de vos données et de votre matériel biologique auprès du médecin qui vous suit dans le cadre de cette recherche.
Vous avez la possibilité à tout moment de demander au médecin qui vous suit dans le cadre de la recherche, la destruction de ces prélèvements biologiques ou de vous opposer à toute utilisation ultérieure.
Si vous décidiez de ne pas participer à cette recherche, vous recevriez vraisemblablement le même traitement standard par chimiothérapie d’induction (« 3+7 »), puis après la fin de votre participation à l’étude vous recevriez les mêmes traitements : cycles de chimiothérapie de consolidation et /ou greffe allogénique de cellules souches hématopoïétiques.
Cependant, l’efficacité du traitement standard de la LAM est inconstante, justifiant la recherche de nouvelles approches comme l’addition de sulfasalazine à la chimiothérapie standard d’induction, qui permettrait d’améliorer le taux de rémissions profondes des LAM, et ainsi, nous l’espérons, prolonger la survie voire augmenter le taux de guérison.
Arrêt prématuré de la recherche :
- Votre médecin pourra décider à tout moment de l’arrêt de votre participation ; il vous en expliquera les raisons.
- Si vous-même décidiez de retirer votre consentement à la recherche, le traitement par sulfasalazine, s’il était encore en cours, serait arrêté et vous ne seriez plus suivi dans le cadre de cette étude. Aucune information nouvelle vous concernant ne pourrait plus être recueillies dans le cadre de cette recherche.
Dans le cadre de la recherche à laquelle il vous est proposé de participer, un traitement de vos données personnelles va être mis en oeuvre par l’AP-HP, promoteur de la recherche, et responsable de traitement, pour permettre d’en analyser les résultats.
Ce traitement est nécessaire à la réalisation de la recherche qui répond à la mission d’intérêt public dont est investie l’AP-HP en tant qu’établissement public de santé hospitalo-universitaire.
A cette fin, les données médicales vous concernant, seront transmises au Promoteur ou aux personnes ou partenaires agissant pour son compte, en France ou à l’étranger. Ces données seront identifiées par un numéro d’enregistrement. Ces données pourront également, dans des conditions assurant leur confidentialité, être transmises aux autorités de santé françaises.
Les données médicales vous concernant pouvant documenter un dossier auprès des autorités compétentes portant sur le médicament évalué dans cette recherche, pourront être transmises à un industriel afin qu’un plus grand nombre de patients puissent bénéficier des résultats de la recherche. Cette transmission sera faite dans les conditions assurant leur confidentialité.
Vos données pourront être utilisées pour des recherches ultérieures ou des analyses complémentaires à la présente recherche en collaboration avec des partenaires privés ou publics, en France ou à l’étranger, dans des conditions assurant leur confidentialité et le même niveau de protection que la législation européenne.
Vous pouvez vous opposer à tout moment à l’utilisation ultérieure de vos données auprès du médecin qui vous suit dans le cadre de cette recherche.
Vos données ne seront conservées que pour une durée strictement nécessaire et proportionnée à la finalité de la recherche. Elles seront conservées dans les systèmes d’information du responsable de traitement jusqu’à dix ans après la dernière publication des résultats de la recherche.
Vos données seront ensuite archivées selon la réglementation en vigueur.
Le fichier informatique utilisé pour cette recherche est mis en oeuvre conformément à la règlementation française (loi « Informatique et Libertés » modifiée) et européenne (Règlement Général sur la Protection des Données -RGPD). Vous disposez d’un droit d’accès, de rectification, de limitation et d’opposition au traitement des données couvertes par le secret professionnel utilisées dans le cadre de cette recherche. Ces droits s’exercent auprès du médecin en charge de la recherche qui seul connaît votre identité (identifié en première page du présent document).
Si vous décidez d’arrêter de participer à la recherche, les données recueillies précédemment à cet arrêt seront utilisées conformément à la réglementation, et exclusivement pour les objectifs de cette recherche. En effet, leur effacement serait susceptible de compromettre la validité des résultats de la recherche. Dans ce cas, vos données ne seront absolument pas utilisées ultérieurement ou pour une autre recherche.
En cas de difficultés dans l’exercice de vos droits, vous pouvez saisir le Délégué à la Protection des données de l’AP-HP à l’adresse suivante : protection.donnees.dsi@aphp.fr, qui pourra notamment vous expliquer les voies de recours dont vous disposez auprès de la CNIL. Vous pouvez également exercer votre droit à réclamation directement auprès de la CNIL (pour plus d’informations à ce sujet, rendez-vous sur le site www.cnil.fr ).
L’AP-HP a pris toutes les mesures pour mener cette recherche conformément aux dispositions du Code de la Santé Publique applicables aux recherches impliquant la personne humaine.
L’AP-HP a souscrit une assurance (N° 0100518814033 220037) garantissant sa responsabilité civile et celle de tout intervenant auprès de la compagnie HDI–GERLING par l’intermédiaire de BIOMEDICINSURE dont l’adresse est Parc d’Innovation Bretagne Sud C.P.142 56038 Vannes Cedex.
L’AP-HP a obtenu l’avis favorable du Comité de Protection des Personnes Sud-Méditerranée II pour cette recherche le 07/07/2022 et une autorisation de l’Agence Nationale de Sécurité du Médicament et des produits de santé (ANSM) le 10/08/2022.
Votre participation à cette recherche est entièrement libre et volontaire. Votre décision n’entraînera aucun préjudice sur la qualité des soins et des traitements que vous êtes en droit d’attendre.
Avant d’accepter de participer à cette recherche, vous bénéficierez d’un examen médical adapté, dont les résultats vous seront communiqués.
Vous pourrez tout au long de la recherche demander des informations concernant votre santé ainsi que des explications sur le déroulement de la recherche au médecin qui vous suit.Vous pouvez vous retirer à tout moment de la recherche sans justification, sans conséquence sur la suite de votre traitement ni la qualité des soins qui vous seront fournis et sans conséquence sur la relation avec votre médecin. A l’issue de ce retrait, vous pourrez être suivi par la même équipe médicale. Dans ce cas, les données collectées jusqu’au retrait seront utilisées pour l’analyse des résultats de la recherche.
Votre dossier médical restera confidentiel et ne pourra être consulté que sous la responsabilité du médecin s’occupant de votre traitement ainsi que par les autorités de santé et par des personnes dûment mandatées par l’AP-HP pour la recherche et soumises au secret professionnel
A l’issue de la recherche et après analyse des données relatives à cette recherche, vous pourrez être informé(e) des résultats globaux en le demandant au médecin qui vous suit dans le cadre de cette recherche.
Vous pouvez également accéder directement ou par l’intermédiaire d’un médecin de votre choix à l’ensemble de vos données médicales en application des dispositions de l’article L 1111-7 du Code de la Santé Publique.
Après avoir lu toutes ces informations, discuté tous les aspects avec votre médecin et après avoir bénéficié d’un temps de réflexion d’au plus 7 jours, si vous acceptez de participer à la recherche vous devrez signer et dater le formulaire de consentement éclairé se trouvant à la fin de ce document.
Etude eTHEMA
Cette étude consiste à mettre en place une cohorte clinique et biologique des leucémies et maladies apparentées (LAM, LAL, SMD de haut-risque ou myélofibrose liée à une NMP).
Elle est qualifiée de non-interventionnelle puisqu’elle ne vous soumet à pas de nouveaux médicaments, de dispositifs médicaux ou de nouveaux outils.
Quels sont les projets de recherches faits avec les échantillons inclus dans eThema ?

NEXT-AML
Notre projet s’appuie sur une méthode de criblage pharmacologique innovante et vise d’abord à établir la faisabilité de cette approche en temps réel pour les patients atteints de LAM en rechute.
Voir le projet
NICHE-RESIST
Identification des mécanismes de chimiorésistance induits par le microenvironnement tumoral dans la LAM
- Nos plateformes technologiques
- Diagnostic
- Leucémies aiguës lymphoïdes
- Leucémies aiguës myéloïdes
- Découvrir l’Institut
- Recherche translationnelle
- Notre recherche clinique
- Essais cliniques
- Devenir patient expert
- Être soigné et accompagné
- Notre actualité
- Un nouvel Institut Hospitalo-Universitaire


{kind=link}
Etude DREAM
Dans le cadre des soins habituels de votre maladie ou dans le cadre d’un protocole, votre médecin vous a proposé un traitement reposant sur l’azacitidine, combiné soit avec le venetoclax soit avec l’ivosidenib selon les caractéristiques de votre maladie, dans le but d’obtenir une rémission de la maladie. L’efficacité clinique de ces traitements est établie. Ce sont des traitements de référence internationalement reconnus pour votre maladie. Pour autant, il est établi que, même en cas d’efficacité maximale de la première cure de traitement, quelques cellules leucémiques résiduelles persistent, nécessitant la poursuite des cycles après l’obtention d’une rémission complète.
La recherche proposée dans le cadre de l’étude DREAM a pour objectif de déterminer s’il est possible, à partir de l’étude au laboratoire d’échantillons de sang ou de moelle osseuse, de prédire l’efficacité du traitement, mais également d’améliorer les connaissances biologiques sur la minorité de cellules leucémiques résiduelles, afin d’identifier des pistes d’amélioration de ces traitements.
Pour cela, l’étude DREAM s’appuie sur de nouvelles technologies disponibles au sein de l’Institut de la Leucémie, et qui permettent de cultiver les cellules leucémiques « in vitro » pour déterminer leur réponse aux traitements, ou d’étudier sur un prélèvement de sang ou de moelle osseuse la façon dont les cellules malades réagissent aux traitements (technologie dite de « séquençage sur cellules uniques »).
Pour répondre à la question posée dans la recherche, cette étude bicentrique à risques et contraintes minimes a prévu d’inclure 120 personnes nouvellement prises en charge dans les services de l’hôpital St Louis et de l’Hôpital Avicenne pour une LAM.
Avant d’initier la première cure de traitement, puis pendant la période d’administration de cette première cure (4 à 10 jours), et enfin à l’évaluation de la réponse au premier et au 6ème cycles (en règle générale, entre 21 et 56 jours après l’initiation du cycle), votre médecin va être amené à prescrire à intervalles réguliers différentes analyses biologiques (cellulaires, moléculaires, protéiques et microbiologiques) du sang et de la moelle osseuse afin de mieux caractériser la maladie, de suivre son évolution pendant le traitement, et de surveiller les conséquences de la maladie et de son traitement sur votre santé.
Dans la recherche proposée, nous allons conduire des études biologiques sur un volume de sang ou de moelle osseuse supplémentaire à certains moments clés du traitement :
Cette étude vise à valider un test de réponse aux médicaments in vitro comme « biomarqueur » de réponse au traitement. Elle a aussi pour objectif d’affiner la connaissance des conséquences moléculaires de l’action de ces médicaments sur les cellules leucémiques, et notamment de déterminer l’hétérogénéité de réponse d’une cellule à l’autre. Elle est donc conduite purement à visée de recherche et n’entraîne aucune modification des traitements que votre médecin vous administrera. Les résultats de ces études biologiques seront obtenus de façon différée par rapport à votre prise en charge médicale et n’auront aucune incidence sur celle-ci.
Ces analyses supplémentaires peuvent nécessiter qu’un maximum de 6 tubes de sang supplémentaires (30 ml) soient prélevés, le plus souvent au cours d’une prise de sang déjà prévue pour votre surveillance. Il est possible que le respect du calendrier des prélèvements de l’étude conduise à la réalisation d’un prélèvement sanguin supplémentaire. De même, 2 mL de moelle osseuse supplémentaires seront prélevés au cours d’une ponction de moelle osseuse effectuée à l’initiation du traitement, en cours de premier cycle, puis lors de la réévaluation au terme du premier cycle. S’il est habituel de pratiquer une ponction de moelle osseuse après le premier cycle de traitement, une ponction intermédiaire supplémentaire sera réalisée autour du 7ème jour de traitement dans le cadre de cette étude. Celle-ci sera déterminante pour permettre aux médecins-chercheurs de comprendre le mécanisme d’action des traitements, afin d’en améliorer à terme l’efficacité.
Enfin, si vous y consentez spécifiquement, votre médecin réalisera, au lieu d’une simple ponction-aspiration de moelle osseuse avant l’initiation du traitement, une ponction-biopsie sous anesthésie locale. Cet examen, qui fait partie des examens de routine de certaines maladies du sang et de la moelle osseuse, est en général pratiqué en cas d’échec de la ponction-aspiration. Avec votre consentement spécifique, cet examen sera pratiqué d’emblée, et le cas échéant lors de la réévaluation. Il permettra, en plus des informations diagnostiques usuellement obtenues avec une ponction-aspiration de disposer d’informations sur leur organisation spatiale que seule peut fournir une ponction-biopsie. De nouvelles technologies (transcriptomique spatiale) permettront aux médecins-chercheurs de mieux comprendre comment sont réparties les cellules malades dans la moelle osseuse, et avec quelles cellules normales du corps (cellules du système immunitaire, cellules de soutien dites « stromales ») elles interagissent. Ceci permettra à terme d’élaborer de nouvelles pistes de traitement.
La durée prévisionnelle de la recherche est de 38 mois et votre participation sera de 56 jours. Après la signature de votre consentement, le déroulement de la recherche sera le suivant :
– un prélèvement de moelle osseuse de 2 mL, au cours du myélogramme d’évaluation initiale de la maladie.
– Optionnellement, si vous y avez spécifiquement consenti, ce myélogramme (ponction-aspiration) sera remplacé par une ponction-biopsie (« biopsie ostéo-médullaire ») sous anesthésie locale,
– un prélèvement sanguin de 30 mL sera réalisé avant tout traitement anti-leucémique, au mieux le même jour que le myélogramme initial.
– Le soir du Jour 1 du premier cycle, entre 6 et 12 heures après l’heure d’administration de la 1ère prise de venetoclax (ou d’ivosidenib) (« bilan de lyse »),
– Le 2ème jour du premier cycle, avant la 2ème prise de venetoclax (ou d’ivosidenib),
– Le jour de l’évaluation de la rémission (entre le 21ème et le 56ème jour après le début du premier cycle).
– Autour du 7ème jour du cycle (entre le 4ème et le 10ème jour)
– Au cours du myélogramme d’évaluation du premier et du 6ème cycle (entre le 21ème et le 56ème jour après le début du cycle).
– Optionnellement, si vous y avez spécifiquement consenti, ce myélogramme (ponction-aspiration) sera remplacé par une ponction-biopsie (« biopsie ostéo-médullaire ») sous anesthésie locale,
En participant à cette recherche, vous contribuerez à une meilleure compréhension du mode d’action des traitements dans les leucémies.
Dans le cadre de cette recherche académique, aucune compensation financière n’est prévue en contre partie de votre participation.
Les contraintes de cette étude sont liées à la nécessité de réaliser les prélèvements de l’étude à heure ou jour fixe, et de rendre impératif la réalisation du myélogramme de réévaluation intermédiaire. Ces contraintes sont minimes car les prélèvements veineux peuvent être réalisés le plus souvent au cours des bilans sanguins requis par la prise en charge usuelle de votre maladie, sans nécessité de ponction veineuse supplémentaire, et que l’ajout d’un myélogramme d’évaluation intermédiaire n’a aucune incidence sur la prise en charge médicale. Les prélèvements sanguins et ostéo-médullaires peuvent entrainer une sensation désagréable transitoire au moment de la ponction, ainsi qu’un hématome au point de ponction.
Si vous acceptez de participer, vous devrez respecter les points suivants :
Après avoir été prélevés, les échantillons de sang et de moelle vont être fractionnés en plasma, cellules et coupes tissulaires (pour les biopsies). Une partie de ces échantillons seront cryoconservés dans l’unité INSERM U944 à l’Hôpital Saint-Louis (Institut de Recherche Saint-Louis) pour une utilisation différée. Cette collection biologique sera déclarée au Ministère de la Recherche et à l’Agence Régionale de Santé (Article L. 1243-3 du Code de la Santé Publique). Une autre sera utilisée directement pour des analyses moléculaires et/ou cellulaires (séquençage d’ARN et/ou d’ADN sur cellules uniques, cytométrie en flux, dosage de protéines plasmatiques). La plupart de ces analyses seront conduites sur le site de l’Hôpital Saint-Louis (Institut de Recherche Saint-Louis). Néanmoins, il est possible que certaines analyses spécifiques à partir des échantillons biologiques ou de leurs dérivés (ARN, ADN, protéines, cellules, etc.) soient conduites par des partenaires académiques extérieurs, en France, en Europe ou ailleurs dans le monde, ou soient soumises à des prestations de service à des entreprises de biotechnologies en France, en Europe ou ailleurs dans le monde. Le cas échéant, ces envois seront conditionnés par l’obtention d’une autorisation d’export d’échantillons biologiques issus de la personne humaine émises par le Ministère de la Recherche, en accord avec les Articles R.1235-7 et suivants du Code de la Santé Publique.
Au terme de l’étude, les échantillons non utilisés seront conservés pour des recherches ultérieures sur la pathologie étudiée. En effet, la collection biologique obtenue dans le cadre de cette étude sera unique du fait de la disponibilité de prélèvements obtenus en cours de traitement, et non pas seulement au diagnostic.
Les échantillons conservés et les données anonymisées, seront accessibles aux investigateurs THEMA. Leur accès (cession ou transfert) par des partenaires de recherche académique pourra être sollicité sur la base d’appels à projets annuels au terme de la présente recherche, et les projets scientifiques devront avoir été approuvés par le Comité de Pilotage de l’étude DREAM.
Les échantillons biologiques seront labélisés « biobanque THEMA – DREAM » et stockés pour une durée maximale de 25 ans (20 ans après le terme de la présente recherche) dans l’unité INSERM U944 de l’Institut de Recherche Saint-Louis. Les données brutes issues des analyses biologiques corrélatives seront stockées de façon sécurisée sur le serveur du bâtiment MEARY de l’Hôpital Saint-Louis. Les données secondaires (biomarqueurs) issues de ces recherches seront regroupées au sein du recueil de données de cliniques de l’étude eTHEMA et gérées par l’Unité de Recherche Clinique de l’Hôpital Saint-Louis. Ces données seront conservées pendant 25 ans après le début de l’étude DREAM.
La réalisation de l’étude DREAM prévoit l’analyse des caractéristiques génétiques somatiques des cellules leucémiques, à l’exclusion de toute caractéristique génétique constitutionnelle, c’est-à-dire potentiellement transmissible à la descendance. Si une analyse biologique sur ces échantillons devait proposer l’étude de caractéristiques génétiques transmissibles, un nouveau consentement dédié vous sera soumis.
Vous avez la possibilité à tout moment de demander au médecin qui vous suit dans le cadre de la recherche, la destruction de ces prélèvements biologiques ou de vous opposer à toute utilisation ultérieure.
En cas de non-participation à cette étude, votre traitement demeurera inchangé, de même que la surveillance des analyses sanguines en cours de traitement, l’évaluation de la moelle osseuse au début et à la fin du premier cycle de traitement.
A la fin de votre participation à l’étude DREAM, votre prise en charge médicale continuera de façon inchangée, ainsi que votre participation au registre eTHEMA, sauf si vous mettez également un terme à votre participation à ce registre. Votre médecin pourra décider à tout moment de l’arrêt de votre participation ; il vous en expliquera les raisons.
Dans le cadre de la recherche à laquelle il vous est proposé de participer, un traitement de vos données personnelles va être mis en oeuvre par l’AP-HP, promoteur de la recherche, et responsable de traitement, pour permettre d’en analyser les résultats.
Ce traitement est nécessaire à la réalisation de la recherche qui répond à la mission d’intérêt public dont est investie l’AP-HP en tant qu’établissement public de santé hospitalo-universitaire.
A cette fin, les données médicales vous concernant seront transmises au Promoteur ou aux personnes ou partenaires agissant pour son compte, en France ou à l’étranger. Ces données seront identifiées par un numéro d’enregistrement et labélisées « base de données THEMA – DREAM ». Ces données pourront également, dans des conditions assurant leur confidentialité, être transmises aux autorités de santé françaises.
Outre le promoteur et les investigateurs du centre THEMA, les seuls destinataires de vos données personnelles codées seront les deux sous-traitants suivants :
• Le fournisseur du logiciel de recueil de données, hébergé en France (celui-ci ne pourra ni les diffuser ni les analyser).
• Le service de gestion des données et de biostatistiques de l’étude, qui fait partie du centre THEMA.
Les données médicales vous concernant pouvant documenter un dossier auprès des autorités compétentes pourront être transmises à un industriel afin qu’un plus grand nombre de patients puisse bénéficier des résultats de la recherche. Cette transmission sera faite dans les conditions assurant leur confidentialité.
Vos données pourront être utilisées pour des recherches ultérieures ou des analyses complémentaires à la présente recherche en collaboration avec des partenaires privés ou publics, en France ou à l’étranger, dans des conditions assurant leur confidentialité et le même niveau de protection que la législation européenne.
Vous pouvez vous opposer à tout moment à l’utilisation ultérieure de vos données auprès du médecin qui vous suit dans le cadre de cette recherche.
Vos données ne seront conservées que pour une durée strictement nécessaire et proportionnée à la finalité de la recherche. Elles seront conservées dans les systèmes d’information du responsable de traitement jusqu’à dix ans après la dernière publication des résultats de la recherche.
Vos données seront ensuite archivées selon la réglementation en vigueur.
Le fichier informatique utilisé pour cette recherche est mis en oeuvre conformément à la règlementation française (loi « Informatique et Libertés » modifiée) et européenne (Règlement Général sur la Protection des Données -RGPD). Vous disposez d’un droit d’accès, de rectification, de limitation et d’opposition au traitement des données couvertes par le secret professionnel utilisées dans le cadre de cette recherche. Ces droits s’exercent auprès du médecin en charge de la recherche qui seul connaît votre identité (identifié en première page du présent document).
Si vous décidez d’arrêter de participer à la recherche, les données recueillies précédemment à cet arrêt seront utilisées conformément à la réglementation, et exclusivement pour les objectifs de cette recherche. En effet, leur effacement serait susceptible de compromettre la validité des résultats de la recherche. Dans ce cas, vos données ne seront absolument pas utilisées ultérieurement ou pour une autre recherche.
En cas de difficultés dans l’exercice de vos droits, vous pouvez saisir le Délégué à la Protection des données de l’AP-HP à l’adresse suivante : protection.donnees.dsi@aphp.fr, qui pourra notamment vous expliquer les voies de recours dont vous disposez auprès de la CNIL. Vous pouvez également exercer votre droit à réclamation directement auprès de la CNIL (pour plus d’informations à ce sujet, rendez-vous sur le site www.cnil.fr ).
L’AP-HP a pris toutes les mesures pour mener cette recherche conformément aux dispositions du Code de la Santé Publique applicables aux recherches impliquant la personne humaine.
L’AP-HP a souscrit une assurance (0100518814033 230094) garantissant sa responsabilité civile et celle de tout intervenant auprès de la compagnie HDI–GERLING par l’intermédiaire de BIOMEDICINSURE dont l’adresse est Parc d’Innovation Bretagne Sud C.P.142 56038 Vannes Cedex.
L’AP-HP a obtenu l’avis favorable du Comité de Protection des Personnes pour cette recherche Comité de protection des personnes Ile de France III le 12/09/2023.
Votre participation à cette recherche est entièrement libre et volontaire. Votre décision n’entraînera aucun préjudice sur la qualité des soins et des traitements que vous êtes en droit d’attendre.
Vous pourrez tout au long de la recherche demander des informations concernant votre santé ainsi que des explications sur le déroulement de la recherche au médecin qui vous suit.
Vous pouvez vous retirer à tout moment de la recherche sans justification, sans conséquence sur la suite de votre traitement ni la qualité des soins qui vous seront fournis et sans conséquence sur la relation avec votre médecin. A l’issue de ce retrait, vous pourrez être suivi par la même équipe médicale. Dans ce cas, les données collectées jusqu’au retrait seront utilisées pour l’analyse des résultats de la recherche.
Vous avez la possibilité à tout moment de demander au médecin qui vous suit dans le cadre de la recherche, la destruction de ces prélèvements biologiques ou de vous opposer à toute utilisation ultérieure.
Votre dossier médical restera confidentiel et ne pourra être consulté que sous la responsabilité du médecin s’occupant de votre traitement ainsi que par les autorités de santé et par des personnes dûment mandatées par l’AP-HP pour la recherche et soumises au secret professionnel.
A l’issue de la recherche et après analyse des données relatives à cette recherche, vous pourrez être informé(e) des résultats globaux en le demandant au médecin qui vous suit dans le cadre de cette recherche
Vous pouvez également accéder directement ou par l’intermédiaire d’un médecin de votre choix à l’ensemble de vos données médicales en application des dispositions de l’article L 1111-7 du Code de la Santé Publique.
Après avoir lu toutes ces informations, discuté tous les aspects avec votre médecin et après avoir bénéficié d’un temps de réflexion, si vous acceptez de participer à la recherche vous devrez signer et dater le formulaire de consentement éclairé se trouvant à la fin de ce document.